





Arzneimittelbehörden zunehmend abhängig von Pharmagebühren (2)
Im ersten Teil dieses Artikels ging es um die Finanzierung wichtiger Arzneimittelbehörden. Die australische Wissenschaftsjournalistin Maryanne Demasi stellt in einem Artikel im «British Medical Journal» einen Zusammenhang her zwischen den finanziellen Abhängigkeiten und der Geschwindigkeit und dem Wohlwollen, mit denen die Behörden Medikamente zulassen. Sie weist auch darauf hin, dass neue Medikamente immer öfter in einem beschleunigten oder erleichterten Zulassungsverfahren das OK erhalten würden.
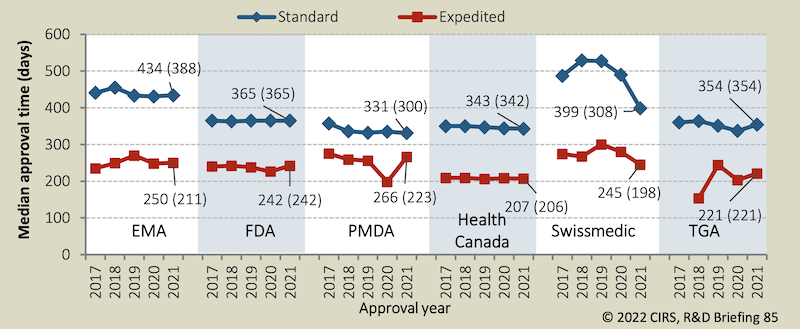
FDA lässt Medikamente am schnellsten zu
Pharmakonzerne kritisierten in der Vergangenheit wiederholt, dass die Zulassungsbehörden zu langsam seien. Einen Ländervergleich liefert alljährlich das «Centre for Innovation in Regulatory Science» in seinem Bericht.
Spitzenreiter beim schnellen Zulassen von Medikamenten ist demnach die US-Behörde FDA: Sie liess in den letzten Jahren etwa zwei von drei neuen Arzneimitteln per Schnellverfahren zu. Für die Hersteller bedeutete dies, dass ihr Medikament im Mittel rund 120 Tage früher auf den Markt kam. Anstatt wie sonst rund ein Jahr dauerte der beschleunigte FDA-Begutachtungsprozess etwa 240 Tage.
Swissmedic prüfte letztes Jahr von 25 von 45 neu zugelassenen Humanarzneimitteln in beschleunigten oder vereinfachten Zulassungsverfahren (siehe Tabelle hier).
Während der Pandemie kam es zu Rekorden: Für die Prüfung und Zulassung des Wirkstoffs Remdesivir brauchte die schnellste von sechs westlichen Behörden im Jahr 2020 nur gerade drei Tage, die «langsamste» 148 Tage. Swissmedic beispielsweise benötigte dank eines «rollenden» Begutachtungsprozesses nur 60 Tage bis zur befristeten Zulassung des Moderna-Impfstoffs. Auch Anträge für Qualitätsänderungen der Covid-19-Impfstoffe, beispielsweise zur Haltbarkeit, bearbeitete Swissmedic zügig: Insgesamt 74 solche Anträge im Jahr 2021 wurden laut dem Geschäftsbericht jeweils innerhalb weniger Tage begutachtet.
Wird die Frist überschritten, schmälert das die Einnahmen
Dank der beschleunigten Zulassungen erhalten Kranke die Medikamente schneller. Doch das birgt auch Risiken: Rasch zugelassene Medikamente mussten öfter aus Sicherheitsgründen wieder vom Markt genommen werden oder wurden später mit Warnungen versehen, gibt Demasi zu bedenken.
Bei den beschleunigten Verfahren müssen die Prüfer kurze Fristen einhalten. Joel Lexchin, ein Experte für Arzneimittelzulassung von der York University in Toronto nennt gegenüber dem «BMJ» einen möglichen Grund, weshalb von der FDA kurz vor Ablauf der Frist zugelassenen Medikamente zu mehr Sicherheitsproblemen geführt haben könnten: Die FDA-Prüfer hätten Angst gehabt, die Frist zu überschreiten und damit Einnahmen der FDA zu gefährden. Von den Einnahmen hängen indirekt Arbeitsplätze bei der FDA ab.
Behörde gibt sich mit vermuteter Wirkung zufrieden
Laut dem renommierten Pharmakologen Aaron Kesselheim sei bei Wirkstoffen, die in beschleunigten Verfahren zugelassen würden, die Messlatte beim Wirksamkeitsnachweis im Allgemeinen tiefer angesetzt. Kesselheim war Mitglied in einem Beratungsausschuss der FDA und warf dort zusammen mit zwei Kollegen letztes Jahr das Handtuch, als die FDA Aducanumab in einem beschleunigten Zulassungsverfahren gegen die Alzheimer-Krankheit zuliess – obwohl der Ausschuss fast einstimmig gegen und niemand im Gremium für diese Zulassung votiert hatte.
Den Beweis, dass Aducanumab wirklich nennenswert gegen Alzheimer hilft, durfte der Hersteller damals schuldig bleiben. Die FDA gab sich damit zufrieden, dass das extrem teure Aducanumab bestimmte Ablagerungen im Gehirn vermindern kann, die mit Alzheimer in Verbindung gebracht werden – ein sogenannter Surrogatparameter. Bereits im Jahr 2017 liess die FDA etwa die Hälfte aller neuen Humanarzneimittel aufgrund von solchen «Ersatzparametern» zu.
«Surrogat-Endpunkte»
«Wenn wir zum Nutzen von Medikamenten […] recherchieren, begegnet uns dieses Phänomen sehr häufig: Studien untersuchen oft gar nicht das, was für die Patientinnen und Patienten wichtig ist, nämlich zum Beispiel, ob sie durch das Medikament länger leben, weniger Beschwerden haben oder sich ihre Lebensqualität verbessert. Stattdessen messen die Forschungsteams Blutwerte, die Knochendichte oder Veränderungen der Grösse eines Krebsgeschwürs. Das Problem: Es ist sehr häufig nicht klar, ob sich positive Veränderungen bei diesen Messwerten tatsächlich auch in dem niederschlagen, was für Patientinnen und Patienten wirklich zählt», stand 2019 in einem Artikel in der Zeitschrift «Gute Pillen Schlechte Pillen» (GPSP).
Insbesondere bei Medikamenten, die im Schnellverfahren zugelassen werden, beruht die Zulassung oft auf vorteilhaften Surrogatwerten. Das kann funktionieren und den Kranken eine raschere Behandlung ermöglichen. «GPSP» führt als Beispiel die Medikamente gegen HIV an, die aufgrund von Surrogatwerten rasch zugelassen wurden. Sie reduzierten die Anzahl der HI-Viren im Blut. Später zeigte sich dann, dass diese Medikamente auch das Fortschreiten von Aids bremsen.
Immer wieder passiert aber auch das Gegenteil: Ein Medikament wird im Schnellverfahren aufgrund von Surrogat-Endpunkten zugelassen – und Jahre später stellt sich heraus, dass es nicht hilft oder sogar schadet.
So geschehen in der Vergangenheit zum Beispiel
- beim Diabetesmedikament Rosiglitazon: Es senkte zwar wirksam den Blutzucker, führte aber bei den damit Behandelten nicht wie erhofft zu weniger, sondern zu mehr Herzinfarkten. 2010 wurde es vom Markt genommen.
- beim Cholesterinsenker Clofibrat: Er senkte zwar das Cholesterin, führte aber zu mehr anstatt zu weniger Todesfällen.
- bei der Einnahme von Hormonen nach den Wechseljahren: Verschiedene Blutwerte wurden davon zwar günstig beeinflusst, aber die Frauen, die diese Hormone schluckten, bekamen öfter Herzinfarkte, Schlaganfälle, Thrombosen oder Brustkrebs.
- bei verschiedenen Krebsmedikamenten: Zwar verbesserten sich die Surrogat-Endpunkte, aber dies führte «weder zu längerem Überleben noch zu einer besseren Lebensqualität», schreibt «GPSP». Eine Analyse im «European Journal of Cancer» ergab, dass nur zwölf Prozent der Surrogatparameter wirklich eng mit einer längeren Überlebenszeit verknüpft waren. Bei 38 Prozent dagegen sagte der – durch die Behandlung verbesserte – Surrogatwert nicht viel darüber aus, ob die Kranken dank der Therapie tatsächlich länger lebten.
In mehr als 90 Prozent der Fälle entscheidet die Europäische Arzneimittelagentur laut dem «BMJ» gleich wie die FDA. Im Fall von Aducanumab war es anders: Die EMA lehnte die Zulassung im Dezember 2021 wegen Zweifeln am Nutzen sowie Sicherheitsbedenken ab. Dazu trug möglicherweise bei, dass das Zulassungsgesuch bei der EMA erst 115 Tage später als bei der FDA eintraf und die EMA so mehr Studiendaten erhielt.
Hersteller kamen den Auflagen nicht nach
Oft lassen die Arzneimittelbehörden Medikamente zu, verlangen jedoch weitere Studien zur Sicherheit, wenn das Medikament bereits auf dem Markt ist. Als Wissenschaftler fünf bis sechs Jahre später einmal überprüften, ob die Hersteller die Auflagen der FDA erfüllt hatten, stellte sich heraus: Nur 54 Prozent der angeforderten Studien waren bis dahin abgeschlossen worden. 25 Prozent waren im Rückstand oder liefen noch und 20 Prozent dieser verlangten Studien hatten die Hersteller noch nicht einmal begonnen. Beim Diabetesmedikament «Victoza» etwa hatten Tierversuche gezeigt, dass die Substanz Schilddrüsenkrebs hervorrufen kann. Die FDA verpflichtete den Hersteller Novo Nordisk, betroffene Patienten mit Schilddrüsenkrebs in einem Register zu erfassen – doch sieben Jahre später habe es dieses Register noch immer nicht gegeben, berichtete das «New England Journal of Medicine» 2017. Novartis hätte auf Geheiss der FDA nach der Zulassung des Multiple Sklerose Medikaments «Gilenya» noch eine Studie mit einer niedrigeren Dosis machen sollen, um festzustellen, ob diese Dosis wirksam und sicherer ist als die höhere Dosis. Aber mehr als sechs Jahre später sei diese Studie noch immer nicht abgeschlossen gewesen. Novartis hatte derweil Milliarden mit «Gilenya» verdient.
Personalrochaden zwischen Behörden und Pharmafirmen
Ein anderes Problem sind die Personalrochaden zwischen Pharmafirmen und Arzneimittelbehörden: «Eine ‹Drehtür› hat dazu geführt, dass viele Beamte der Behörde für dieselben Unternehmen, die sie reguliert haben, arbeiten oder sie beraten», erklärt Maryanne Demasi im «BMJ». Neun von zehn der ehemaligen FDA-Leiter zwischen 2006 und 2019 arbeiteten nach ihrem Weggang von der Behörde für pharmazeutische Unternehmen oder berieten diese.
Ein prominentes Beispiel ist Stephen Hahn. Er leitete die FDA bis Januar 2021. Unter seiner Führung wurde der mRNA-Impfstoff von Moderna notfallmässig zugelassen. Sechs Monate später wechselte Hahn den Posten und arbeitet nun genau für die Firma, die Moderna hervorgebracht hat. Sein neuer Arbeitgeber «Flagship» ist ein Venture Fund, der biopharmazeutische Unternehmen auf den Weg bringt.
Im Februar 2022 wurde der Herzspezialist Robert Califf zum neuen Leiter der FDA ernannt – ungeachtet dessen, dass er 2,7 Millionen Dollar von einer Life Sciences Firma erhalten hatte und noch 2021 Vorstandsmitglied bei zwei pharmazeutischen Unternehmen war.
Ein anderes Beispiel ist Ian Hudson, der erst Vizedirektor bei der Pharmafirma SmithKline Beecham war, dann zum Leiter der britischen Arzneimittelbehörde MHRA aufstieg und jetzt die Bill und Melinda Gates Stiftung berät sowie für eine Biotech Firma tätig ist.
Ruf nach Gremien, die unabhängig von der Industrie urteilen
Das Fazit des Medikamentenzulassungs-Experten Donald Light: Für Patienten und Ärzte sei es nicht mehr möglich, von den Arzneimittelbehörden neutrale, rigorose Beurteilungen zu erhalten. Die Behörden bräuchten inzwischen selbst einen Überwacher. Er schlägt vor, Non-Profit-Organisationen wie etwa das deutsche «Institut für Wirtschaftlichkeit und Qualität im Gesundheitswesen» (IQWIG) zu schaffen, das Medikamente und Behandlungsmethoden unabhängig von der Industrie und transparent überprüfe.
Was Light nicht erwähnt: Ausgerechnet das international anerkannte IQWIG wurde von der deutschen Politik in der Pandemie praktisch komplett übergangen. Warum, weiss das IQWIG nicht.
Themenbezogene Interessenbindung der Autorin/des Autors
Keine
_____________________
Meinungen in Beiträgen auf Infosperber entsprechen jeweils den persönlichen Einschätzungen der Autorin oder des Autors.





Ein Nebenprodukt dieser sehr interessanten Arbeit: Die Schweizer Zulassungsstelle benötigte bis 2020 durchschnittlich gut 100 Tage länger für eine Standardzulassung. Ob die Qualität der Arbeit auch entsprechend höher war als bei den Behörden der anderen Länder, ist offen.
Die Schweiz musste auch bei der Zulassung etwas verzögert vorgehen, so dass das Erstzulassungsdatum im EWG-Land Liechtenstein nicht die Zeit der möglichen «Patentverlängerung» in der EU kürzen würde.
Schliesslich hat sich die Schweiz mit der Pharma arrangiert indem das offizielle Zulassungsdatum für Liechtenstein ein Jahr nach dem Datum für die Schweiz festgesetzt wird